How to Use CHITRA
Step-by-step guide to using CHITRA and exploring example datasets
HOW TO USE CHITRA?
Below are the step-by-step instructions, accompanied by pictorial representations, to ensure a seamless experience with CHITRA.
Step 1: Getting Started
- On the landing page, click the “Get Started” button to begin.
Step 2: Uploading Files or Viewing Example Data
- On the next page, you can either:
- Upload Files
- View Example Datasets
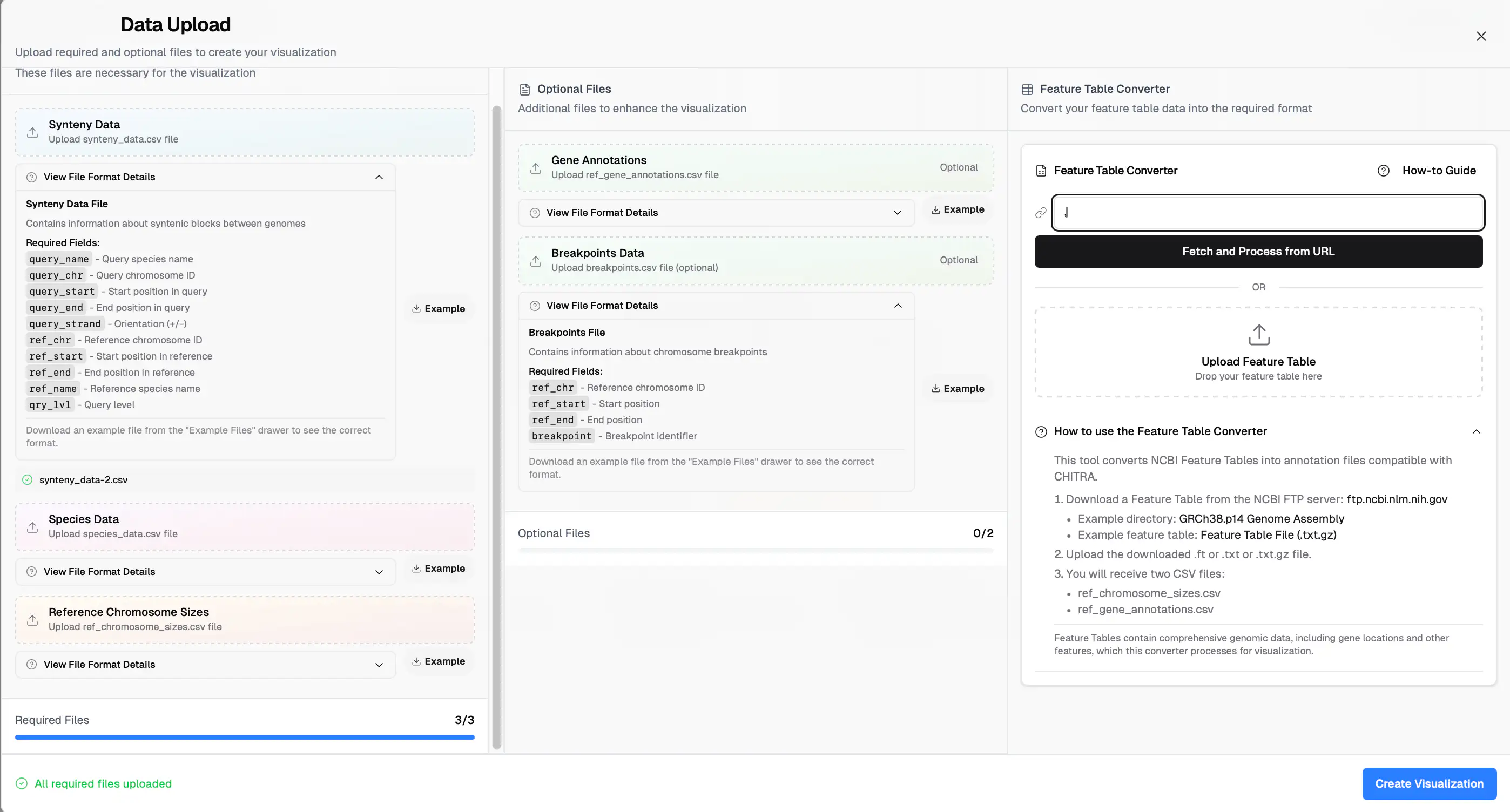
2.1.1 File Upload Instructions
Mandatory Input Files:
- Synteny Data
- Genome Data
- Reference Chromosome Size
Optional Input Files:
- Gene Annotations
- Breakpoint Data
To upload files:
Click the “Upload Data” button. A popup window will appear where you can browse and upload the required input files.
Note: All input files should be in CSV format
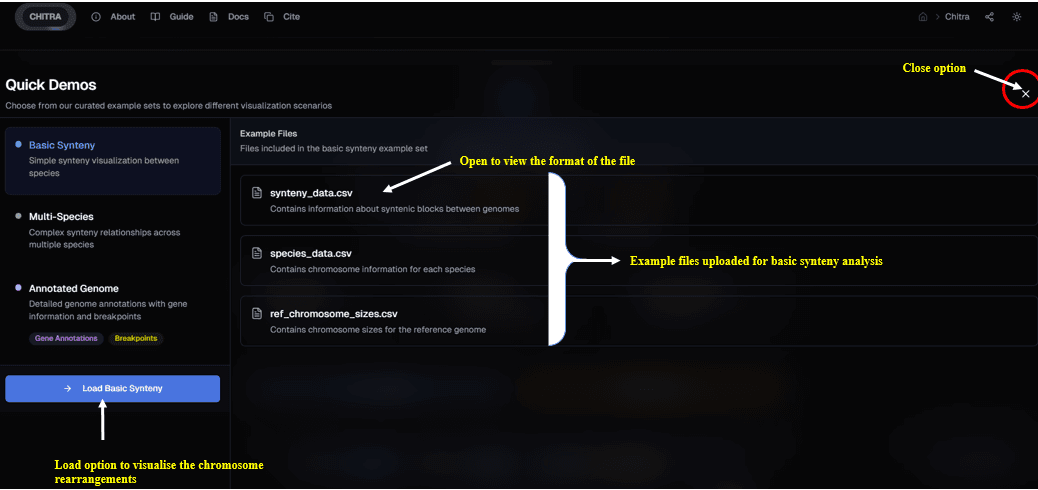
2.1.2 Using Example Datasets
CHITRA includes pre-loaded example datasets to help users explore its functionality. Three types of example datasets are available:
1. Basic Synteny
Visualize synteny blocks among three genomes.
Input files required:
2. Multiple Synteny
Compare synteny blocks among multiple genomes. Uses the same files as Basic Synteny.
3. Annotated Genome
Analyze annotated genomes using real datasets. Uses the same files as Basic Synteny, plus these additional files:
Available Datasets
Three types of example datasets are available:
1. Basic Synteny
- Simple synteny visualization between genome.
- Ideal for understanding the fundamental concepts of synteny blocks and chromosome mapping.
2. Multi-Genome
- Complex synteny relationships across multiple genome.
- Allows comparison of genomic architecture across diverse organisms.
- Includes data for multiple pairwise comparisons.
3. Annotated Genome
- Detailed genome annotations with gene information and breakpoints.
- Includes gene features, locus tags, and symbols.
- Provides breakpoint data for structural variation analysis.
[!NOTE] You can preview and download the example data tables directly from the example files drawer below.
Exploring Example Data
- Select any of the three options in the drawer to access the files.
- You can preview the data or download the files directly.
- Click "Load" to automatically populate the visualization with the selected dataset.
Feature Table Converter
This tool converts NCBI Feature Tables into annotation files compatible with CHITRA.
How to use the Feature Table Converter
-
Download a Feature Table
- Get the file from the NCBI FTP server.
- Example directory: GRCh38.p14 Genome Assembly
- Example file: Feature Table File (.txt.gz)
-
Upload the File
- Upload the downloaded
.ft,.txt, or.txt.gzfile to the converter.
- Upload the downloaded
-
Download Converted Files
- You will receive two CSV files compatible with CHITRA:
ref_chromosome_sizes.csv: Contains the size of each chromosome.ref_gene_annotations.csv: Contains detailed gene annotation data.
- You will receive two CSV files compatible with CHITRA:
Note: Feature Tables contain comprehensive genomic data, including gene locations and other features, which this converter processes for visualization.
Data Preparation & External Tool Integration
To assist users in transitioning from standard bioinformatics tool outputs to CHITRA, we provide the following mapping guidance.
Mandatory CSV Schema Definitions
Before preparing your data, ensure your CSV files follow these column naming conventions:
| File | Required Columns | Description |
|---|---|---|
| Synteny Data | ref_chr, ref_start, ref_end, query_chr, query_start, query_end, query_strand, ref_name, query_name | Coordinates and orientation (+/-) of syntenic blocks between the reference and query. |
| Genome Data | genome_name, chr_id, chr_size_bp | Full list of chromosomes and their sizes for all genome involved. |
| Reference Sizes | chromosome, size | Specific sizes for the primary reference genome used for coordinates. |
Mapping from Popular Synteny Callers
If you are using external tools, map their outputs to CHITRA's schema as follows:
| Tool | Primary Output File | Mapping to CHITRA Fields |
|---|---|---|
| SyntenyTracker2 | Natively Compatible | Recommended: Automatically generates CHITRA-ready CSV files. |
| Satsuma2 | satsuma_summary.chained.out | Ref: Col 1-3 → ref_chr, ref_start, ref_endQuery: Col 4-6 → query_chr, query_start, query_end |
| SyRI | syri.out | Ref: Col 1-3 → ref_chr, ref_start, ref_endQuery: Col 6-8 → query_chr, query_start, query_end |
| Minimap2 | .paf | Ref: Col 6, 8, 9 → ref_chr, ref_start, ref_endQuery: Col 1, 3, 4 → query_chr, query_start, query_end |
| MCScanX | .collinearity | Manual or script extraction of block boundaries required. |
SyntenyTracker2: The Preferred Workflow
For a seamless experience, we recommend SyntenyTracker2 as the companion tool for data preparation. It is designed to modernize modular synteny pipelines and simplify the path to visualization by generating data with exact genomic coordinates.
Installation via Conda:
conda install jitendralab::syntenytracker2By using SyntenyTracker2, you can generate all mandatory input files for CHITRA in one step, ensuring high reproducibility and significantly reducing the data preparation burden.
Working with MCScanX Data
MCScanX is a popular comparative genomics toolkit used to detect syntenic and collinear gene blocks from raw genome annotation and homology data.
Typical MCScanX Workflow:
- Generate all-vs-all BLASTP results between your genomes.
- Prepare a GFF/BED file containing your gene coordinates.
- Run MCScanX using these inputs to produce a
.collinearityfile for downstream analysis.
[!WARNING] Important Limitation regarding MCScanX: MCScanX predicts collinearity by connecting individual homologous genes based on e-values (e.g., matching
chr1_gene001tochr3_gene221), rather than exporting the exact start/end boundary coordinates of a contiguous physical DNA block. Since CHITRA currently requires exact boundary mappings (like those produced by Satsuma or SyntenyTracker) to render physical chromosome ribbons, MCScanX.collinearityoutputs currently require you to run a custom intermediate script to convert those gene-to-gene links into standard block coordinates before uploading to CHITRA.
Future Support: In a future release, CHITRA plans to directly support parsing .collinearity output files. We intend to automatically infer and group these connected lower-e-value threshold genes to bridge and generate broader syntenic regions between chromosomes directly within the application.
Raw Data Viewer — Column Reference
The Raw Data Viewer (accessible via the View Data button in the toolbar) displays the loaded data across five tabs. Each column header has an inline ℹ tooltip and a Column Guide button per tab. The full definitions are listed below.
[!NOTE] All positional values (start, end, size) are in base pairs (bp) unless stated otherwise. Columns marked (optional) may show
N/Aif not provided in the input file.
Synteny Table
Displays pairwise synteny block coordinates between the reference and query genomes.
| Column | Field in CSV | Description |
|---|---|---|
| S.No | — | Sequential row number (auto-generated) |
| Query Name | query_name | Name of the query genome being compared against the reference |
| Query Chr | query_chr | Chromosome identifier in the query genome (e.g., chr1) |
| Query Start | query_start | Start position of the synteny block on the query chromosome (bp) |
| Query End | query_end | End position of the synteny block on the query chromosome (bp) |
| Strand | query_strand | Alignment orientation: + = forward/syntenic, − = reverse/inverted |
| Ref Chr | ref_chr | Chromosome identifier in the reference genome |
| Ref Start | ref_start | Start position of the synteny block on the reference chromosome (bp) |
| Ref End | ref_end | End position of the synteny block on the reference chromosome (bp) |
| Ref Genome | ref_name | Name of the reference genome |
| Symbol | symbol | Gene symbol associated with this block, if annotated (optional) |
| Class | class | Functional or structural class of the block (e.g., gene, repeat) (optional) |
| Gene ID | GeneID | NCBI Gene identifier for the annotated gene (optional) |
Genome Table
Lists all chromosomes and their sizes for every genome loaded into CHITRA.
| Column | Field in CSV | Description |
|---|---|---|
| S.No | — | Sequential row number (auto-generated) |
| Genome Name | genome_name | Name of the species or genome assembly |
| Chr ID | chr_id | Chromosome identifier within this genome (e.g., chr1, chrX) |
| Type | chr_type | Chromosome type, e.g., autosome or sex chromosome (optional) |
| Size (bp) | chr_size_bp | Total length of the chromosome in base pairs |
| Centromere Start | centromere_start | Start position of the centromere region (bp) (optional) — when provided, CHITRA renders the characteristic constriction notch on the chromosome ideogram |
| Centromere End | centromere_end | End position of the centromere region (bp) (optional) — used together with centromere_start to draw the centromere |
[!NOTE]
centromere_startandcentromere_endare optional. If omitted, chromosomes are drawn as plain rectangular bars without a centromere marker. Providing them enables metacentric/submetacentric chromosome morphology rendering.
Reference Table
Contains size information specifically for the primary reference genome used as the coordinate base.
| Column | Field in CSV | Description |
|---|---|---|
| S.No | — | Sequential row number (auto-generated) |
| Chromosome | chromosome | Reference chromosome identifier |
| Size (bp) | size | Total length of the reference chromosome in base pairs |
| Centromere Start | centromere_start | Start position of the centromere region (bp) (optional) |
| Centromere End | centromere_end | End position of the centromere region (bp) (optional) |
Genes Table
Gene annotation data for the reference genome (loaded via ref_gene_annotations.csv or the Feature Table Converter).
| Column | Field in CSV | Description |
|---|---|---|
| S.No | — | Sequential row number (auto-generated) |
| Chr | chromosome | Chromosome on which the gene is located |
| Accession | genomic_accession | NCBI genomic accession number for the sequence containing this gene |
| Start | start | Gene start position on the chromosome (bp) |
| End | end | Gene end position on the chromosome (bp) |
| Strand | strand | Gene orientation: + = forward, − = reverse |
| Class | class | Gene biotype or functional class (e.g., protein_coding, pseudogene, ncRNA) |
| Symbol | symbol | Official gene symbol (e.g., BRCA1) (optional) |
| Name | name | Full descriptive name of the gene (optional) |
| Locus Tag | locus_tag | Systematic locus tag identifier assigned by NCBI (optional) |
| Gene ID | GeneID | NCBI Gene identifier (numeric) (optional) |
Breakpoints Table
Chromosomal breakpoint intervals on the reference genome (loaded via breakpoints.csv).
| Column | Field in CSV | Description |
|---|---|---|
| S.No | — | Sequential row number (auto-generated) |
| Ref Chr | ref_chr | Reference chromosome on which the breakpoint is located |
| Start Pos | ref_start | Start position of the breakpoint interval (bp) |
| End Pos | ref_end | End position of the breakpoint interval (bp) |
| Breakpoint Type | breakpoint | Type of chromosomal rearrangement (e.g., inversion, translocation, deletion) |